课程名称:仪器分析 课程编号:072610315
课程类型:(必修/选修)专业必修课
课程总学时: 48 课程总学分:4
开课单位:4 适用专业:化学
一、实验目的与任务
(一)课程的性质
《仪器分析实验》仪器分析实验是高等院校化学化工、轻工、环保、生物技术等有关专业必修的一门化学基础课。本课程是仪器分析课程的重要组成部分,主要介绍了常见分析仪器的使用方法和应用。课程的主要目的是使学生掌握常见分析仪器的基本构造、使用方法及其在分析测试中的应用。培养学生严谨和实事求是的科学态度,利用分析仪器手段解决分析问题和正确处理实验结果、数据等的能力。
(二)课程教学目标基本要求
本课程是学生在学习仪器分析理论课程的基础上,学习和掌握仪器分析的基本原理、基础知识、仪器构造和基本操作技能,并能达到一定的熟练程度,能较好的了解和掌握一般仪器的正确使用和日常维护方法。是一门实践性很强的基础课。本课程的培养目标是使学生在理解、掌握仪器分析的基本原理、基本知识和基本操作技能的基础上,对某一具体的分析对象,能综合应用多门学科知识,逐步达到具有查阅文献资料,选择分析方法,拟定实验步骤,正确评估测定结果的能力,同时也为后继课程的学习打下良好的基础。
二、实验项目基本情况一览表
| 序 号 |
实验项目 |
内容提要 |
学时 |
实验 类型 |
必修 (选修) |
| 1 |
实验一 |
邻二氮菲吸光光度法测定试样中微量铁 |
4 |
综合 |
必修 |
| 2 |
实验二 |
差值光谱法测定废水中的苯酚 |
4 |
综合 |
必修 |
| 3 |
实验三 |
火焰原子吸收光谱法测定水中的钙 |
4 |
综合 |
必修 |
| 4 |
实验四 |
荧光分光光度法测多维葡萄糖中的维生素B2 |
4 |
综合 |
必修 |
| 5 |
实验五 |
荧光分光光度法测荧光素钠的含量 |
4 |
综合 |
必修 |
| 6 |
实验六 |
溴化钾压片法测绘苯甲酸的红外吸收光谱 |
4 |
综合 |
必修 |
| 7 |
实验七 |
水样中pH值的测定 |
4 |
验证 |
必修 |
| 8 |
实验八 |
氟离子选择性电极测定水样中的氟 |
4 |
综合 |
必修 |
| 9 |
实验九 |
库伦滴定法测定水样微量神 |
4 |
综合 |
必修 |
| 10 |
实验十 |
溶出伏安法测定水样中的镉 |
4 |
综合 |
必修 |
| 11 |
实验十一 |
液相色谱法测定污染水样中的甲苯 |
4 |
综合 |
必修 |
| 12 |
实验十二 |
气相色谱法测定酒样中甲醇 |
4 |
设计 |
必修 |
三、实验教学参考书、指导书
1.华中师范大学、东北师范大学、山西师范大学、北京师范大学合编《分析化学实验》高等教育出版社 2001年7月第3版
2.陈培榕 邓勃 《现代仪器分析实验与技术》 清华大学出版社1999年12月第一版
3.张剑荣,戚苓,方惠群 编《仪器分析实验》 科学出版社,1999年第一版
四、说明
本课程与《普通物理》、《高等数学》、《分析化学》等基础主干课和专业基础课联系十分密切,应在这三门先修课程的基础上进行教学。为《有机化学》等化学专业课方面知识的实际运用打下坚实的基础。
五、各实验项目说明
实验一 邻二氮菲吸光光度法测定试样中微量铁
(一)实验目的
1.掌握光度法测定铁的原理及方法。
2.学会VIS-7220分光光度计的正确使用。
3.学习如何选择吸光光度分析的实验条件。
(二)方法原理
邻二氮菲(o-ph)是测定微量铁的较好试剂。在pH=2~9的溶液中,试剂与Fe 2+生成稳定的红色配合物,其lgK形=21.3, 摩尔吸光系数ε= 1.1×104,其反应式如下:
Fe 2+ + 3(o-ph)⇌Fe(o-ph)3
红色配合物的最大吸收峰在510nm波长处。当铁为+3价时,可用盐酸羟胺还原, 本方法的选择性很高,相当于含铁量40倍的Sn 2 +、Al 3+、Ca 2+、Mg 2+、Zn 2+、SiO3 2-,20倍Cr 3+、Mn 2+、V(V)、PO4 3-,5倍Co 2+、Cu 2+等均不干扰测定。
(三)主要仪器及试剂
1. 7220分光光度计,10mL吸量管,50mL比色管8支,1cm比色皿,瓷坩埚,电炉,马弗炉。
2.铁标准溶液:含铁0.1mg/ml。
准确称取0.8634g的NH4Fe(SO4)2·12H2O,置于烧杯中,加入20ml 1:1HCl和少量水,溶解后,定量地转移至1升容量瓶中,以水稀释之刻度,摇匀。
3.邻二氮菲:0.15%(10-3mol/L)新配制的水溶液。
4.盐酸羟胺: 10%水溶液(临用时配制)
5.醋酸钠溶液 1mol/L
6.NaOH 1mol/L
7.HCl 6 mol/L
8.工业盐酸
(四)测定步骤
1.吸收曲线的制作和测量波长的选择:
用吸量管吸取0.0,1.0mL铁标准溶液,分别注入两个50mL比色管中,各加入1mL盐酸羟胺溶液,2mL邻二氮菲,5mL NaAc,用水稀释至刻度,摇匀。放置10min后,用1cm比色皿,以试剂空白(即0.0mL铁标准溶液)为参比溶液,在440~560nm之间,每隔10nm测一次吸光度,在最大吸收峰附近,每隔5nm测定一次吸光度。在坐标纸上,以波长λ为横坐标,吸光度A为纵坐标,绘制A和λ关系的吸收曲线。从吸收曲线上选择测定Fe的适宜波长,一般选用最大吸收波长λmax。
2.标准曲线的制作:
用移液管吸取100μg·mL-1铁标准溶液10mL于100mL容量瓶中,加入2mL 2mol·L -1的HCl,用水稀释至刻度,摇匀。此溶液每毫升含Fe 3+ 10μg。
在6个50mL比色管中,用吸量管分别加入0.0, 2.0, 4.0, 6.0, 8.0, 10.0 mL 10μg·mL -1铁标准溶液,分别加入1mL盐酸羟胺,2mL Phen,5mL NaAc溶液,每加一种试剂后摇匀。然后,用水稀释至刻度,摇匀后放置10 min。用1cm比色皿,以试剂为空白(即0.0mL铁标准溶液),在所选择的波长下,测量各溶液的吸光度。以含铁量为横坐标,吸光度A为纵坐标,绘制标准曲线。用绘制的标准曲线,重新查出相应铁浓度的吸光度,计算Fe 2+-Phen络合物的摩尔吸光系数
3.试样中铁的测定
准确移取5mL样液至50mL容量瓶后,加盐酸羟胺2mL,Phen 2mL,1mol·L -1 NaAc 10mL,加水稀释至刻度,摇匀。测量吸光度A。根据标准曲线求出试样中铁的含量(μg·mL -1)。
实验二紫外差值光谱法测定废水中的微量酚
一、实验目的
1.了解紫外可见分光光度计的使用方法。
2.掌握紫外差值光谱法测定微量酚的基本原理。
二、实验原理
苯酚在紫外区有两个吸收峰,在中性溶液中lmax为210nm和270nm,在碱性溶液中,由于形成酚盐,而使该吸收峰红移至235nm和288nm。所谓差值光谱就是指这两种吸收光谱相减而得到的光谱曲线。实验中只要把苯酚的碱性溶液放在样品光路上,把中性溶液放在参比光路上,即可直接绘出差值光谱。
在苯酚的差值光谱图上,选择288nm为测定波长,在该波长下,溶液的吸光度随苯酚浓度的变化有良好的线性关系,遵循比耳定律,即DA=De×C×L,可用于苯酚的定量分析。差值光谱法用于定量分析,可消除试样中某些杂质的干扰,简化分析过程,实现废水中的微量酚的直接测定。
三、试剂和仪器
试剂:0.1MKOH溶液。0.2500g/L苯酚标准溶液。
仪器:紫外可见分光光度计。1cm厚石英比色吸收池2个。
25ml容量瓶12个
四、实验步骤
1.确定测定波长:以蒸馏水作参比,分别绘制苯酚在中性和碱性溶液中的吸收曲线。然后,将苯酚的中性和碱性溶液分别放置在参比和样品光路中,绘制二者的差值光谱曲线,根据该差值光谱曲线,确定其测定波长。
2.绘制标准曲线:用移液管分别移取苯酚标准溶液1.0ml、1.5ml、2.0ml、2.5ml、3.0ml于5个25ml的容量瓶中,另取同样体积苯酚标准溶液于另5个25ml容量瓶中,分别用水和0.1MKOH稀释至刻度(共需10个25ml容量瓶)。每对容量瓶所对应的溶液浓度分别是10mg/L、15mg/L、20mg/L、25mg/L、30mg/L。每一对苯酚标准溶液中的苯酚浓度相同,只是稀释溶剂不同。在测定波长下,把碱性溶液稀释的标准溶液放在样品光路上,把中性溶液稀释的标准溶液放在参比光路上,测定吸光度差值。
3.测量未知样品中苯酚含量:用移液管分别移取含酚水样10ml于2个25ml容量瓶中,分别用水和0.1MKOH稀释至刻度。在测定波长下,把碱性溶液稀释的待测试样放在样品光路上,把中性溶液稀释的待测试样放在参比光路上,测定吸光度差值。
五、数据处理
1.用实验步骤2中测得的吸光度差值,绘制吸光度—浓度曲线,计算回归方程。
2.用吸光度—浓度曲线或回归方程,计算水样中的苯酚含量(mg/L)。
六、思考题
1. 苯酚的差值光谱图中有235nm和288nm两个吸收峰,为何选288nm作为测定波长?
2.本实验所用的差值光谱法和示差分光光度法有何不同?
附录:TU-1910紫外可见分光光度计操作规程
1.打开仪器电源,仪器开始初始化,一切正常后进入主界面。
2.选择光谱测量,按F1,进行参数设置:
(1) 光度方式:A
(2) 扫描速度:快
(3) 采样间隔:1.0
(4) 波长范围:400~200nm
(5) 纵坐标的范围:0.000~1.000
3.按“RETURN”键,返回光谱测量界面,放入空白样品,按“AUTO ZERO”键进行基线校正。然后,放入待测样品,按“START”键,进行光谱扫描。所得吸收光谱曲线显示在主机屏幕上,按F2,输入阈值,按“确认”,系统将自动检索并显示峰值。据吸收曲线选择最佳测定波长。按F4,可打印谱图。
4.在主界面上,选“定量测定”,按F1,进行参数设置:
(1) 标样法
(2) 测量波长:287nm
(3) 浓度单位:mg/L
5.按F1键,进入标样法参数设置,、
(1) 按“1”,输入标样数:5
(2) 按“2”,标样序号:依次输入1-5#标样的浓度
(3) 在一号池中放入空白样,按“AUTO ZERO”键,进行行空白自动校零。
(4) 按“3”,将样品放入一号池中,按“START”键,测定吸光度。
(5) 重复(4)中的操作,直到5个样品测完。
(6) 按“4”,绘制工作曲线。
6.按“RETURN”键,返回定量测量界面,放入空白样品,按“AUTO ZERO”键进行空白自动校零。然后,放入待测样品,按“START”键,进行水样吸光度测量。界面自动显示测量结果。
7.按“RETURN”键,返回主界面,直接关主机电源。
。
实验三火焰原子吸收光谱法测定水中的钙
(一)实验目的
1.掌握火焰光度法测定钙、镁的方法和火焰光度法正确使用。
2.掌握标准曲线法的原理及应用,熟悉标准曲线法操作技术及数据处理。
(二)实验原理
在使用锐线光源条件下,基态原子蒸汽对共振线的吸收,符合朗伯-比尔定律,即: A=lg(I0/I)=KLN0 (2-2-6)
在试样原子化时,火焰温度低于3000 K时,对大多数元素来讲,原子蒸汽中基态原子的数目实际上十分接近原子总数。在一定实验条件下,待测元素的原子总数目与该元素在试样中的浓度呈正比。用A-c标准曲线法或标准加入法,可以求算出元素的含量。
(三)仪器与试剂
1.仪器 原子吸收分光光度计;钙、镁空心阴极灯。
2.试剂
(1)1.0g.L-1镁标准储备液
(2)1.0g.L-1钙标准储备液
(3)50 mg.L-1镁标准使用液
(4)100 mg.L-1钙标准使用液
(5)MgO(GR);无水CaCO3(GR);HCl(AR)
配制用水均为二次蒸馏水。
(四)实验步骤
1.钙、镁系列标准溶液的配制
配制钙系列标准溶液:2.0,4.0,6.0,8.0,10.0 mg.L-1。
配制镁系列标准溶液:0.1,0.2,0.3,0.4,0.5 mg.L-1。
2.工作条件的设置
吸收线波长 Ca 422.7 nm,Mg 285.2 nm
空心阴极灯电流4 mA
狭缝宽度0.1 mm
原子化器高度6 mm
空气流量4 L.min-1,乙炔气流量1.2 L.min-1
3.钙的测定
(1)用10 mL的移液管吸取自来水样于100 mL容量瓶中,用蒸馏水稀释至刻度,摇匀。
(2)在最佳工作条件下,以蒸馏水为空白,测定钙系列标准溶液和自来水样的吸光度A。
4.镁的测定
(1)用2 mL的吸量管吸取自来水样于100 mL容量瓶中,用蒸馏水稀释至刻度,摇匀。
(2)在最佳工作条件下,以蒸馏水为空白,测定镁系列标准溶液和自来水样的吸光度A。
5.实验结束后,用蒸馏水喷洗原子化系统2 min,按关机程序关机。最后关闭乙炔钢瓶阀门,旋松乙炔稳压阀,关闭空压机和通风机电源。
6.绘制钙、镁的A-c标准曲线,由未知样的西光度Ax,求算出自来水中钙、镁含量(mg.L-1)。或将数据输入微机,按一元线性回归计算程序,计算钙、镁的含量。
(五)注意事项
1.乙炔为易燃易爆气体,必须严格按照操作步骤工作。在点燃乙炔火焰之前,应先开空气,后开乙炔;结束或暂停实验时,应先关乙炔,后关空气。乙炔钢瓶的工作压力,一定要控制在所规定范围内,不得超压工作。必须切记,保障安全。
2.实验结束后,检查仪器是否正常,关闭是否正确。
六、思考题
1.为什么空气、乙炔流量会影响吸光度的大小?
2.为什么要配制钙、镁标准使用溶液?所配制的钙、镁系列标准溶液可以放置到第二天再继续使用吗?为什么?
实验四 荧光光度法测定荧光素含量的测定
一、实验目的
1.掌握荧光分析方法。
2.学会使用荧光分光光度计。
二、实验原理
荧光是光致发光。当物质的分子吸收光以后,从基态跃迁到激发态,为激发态分子,处于激发态的分子通过无辐射去活(释放能量),回到第一电子激发态的最低振动能级,再以发射辐射的形式去活,跃迁回至基态的各个振动能级则发出荧光。
有苯环或具有多个共轭双键体系以及具有刚性平面结构的有机物分子易产生荧光。大多数无机盐金属离子不产生荧光,而一些金属鳌合物能产生很强的荧光。
取代基的性质、溶剂的极性、体系的pH值和温度都会影响荧光体的荧光特性或荧光强度。
1.荧光光谱定量分析原理
溶液的荧光强度F与该溶液对光吸收的程度、溶液中荧光物质的荧光效率及浓度有关:
F=2.303ΦI0bεc
式中Φ为荧光效率,为发射的光子数与吸收的光子数之比;I0为激发光强度;ε为摩尔吸光系数;b为光程长度,c为荧光物质浓度。
当入射光强度一定时,
F=Kc
只有在c较小时上式才适用。即在低浓度时,荧光强度F与溶液荧光物质的浓度c成正比例关系。
荧光分析仪由光源、单色器、液池和检测器等几个部分组成,其结构如图1。
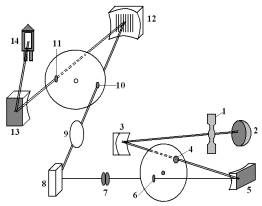
图1荧光分析仪光路图
1-氙灯,2-凸面镜,3-凹面镜,4-入射狭缝,5-激发凹面光栅
6-出射狭缝,7-双透镜,8-样品池,9-透镜,10-入射狭缝
11-出射狭缝,12-发射凹面光栅,13-凹面镜,14-光电倍增管
2.激发光谱
固定荧光最大发射波长,改变激发光波长,测得荧光强度与激发光波长的关系即为激发光谱曲线,由激发光谱曲线可选出最大激发波长。
3.荧光光谱
固定最大激发光波长,测定不同发射波长时的荧光强度,即得到荧光光谱曲线。由荧光光谱曲线可选出最大发射波长。
激发光谱与荧光光谱有镜像关系。
荧光素产生较高的荧光量子效率(约0.85),低浓度时产生强的荧光,在pH=5~10范围内pH与荧光有关,用0.1 mol•L-1 NaOH溶液保证恒定的发光效率。
二、实验主要用品
荧光分光光度计(F96PRO)
容量瓶(50mL)
移液管(5mL)
荧光素、NaOH
三、实验步骤与内容
1.标准溶液配制
荧光素标准溶液I(1.0×10-3mol•L-1):称0.0166克荧光素于小烧杯中,加少量水溶解,转移到50mL容量瓶中;加1moL/L NaOH 5mL,用水稀释至刻度。
荧光素标准溶液II:取1mL荧光素标准溶液I于100mL容量瓶中,加1mol•L-1NaOH 10mL,用水稀释到刻度,配成含荧光素为1×10-5mol•L-1的0.1 mol•L-1NaOH溶液。
2.扫描荧光发射光谱
取荧光素标准溶液II 2mL于50 mL容量并中,加5 mL 1mol•L-1NaOH溶液,用水稀释至刻度。将该溶液装入样品池中,在F96Pro荧光分光光度计上扫描荧光发射光谱,并找出萤光发射波长。
3.荧光素的定量分析
(1)绘制工作曲线
取荧光素标准溶液II 1.0、2.0、3.0、4.0、6.0mL分别于5个50mL容量并中,加5mL 1mol•L-1NaOH,用水稀释至刻度,在F96Pro荧光分光光度计上测量其每个溶液荧光强度。由实验数据可绘制标准曲线。
(2)测定未知样品
以绘制工作曲线的条件测定未知样品的荧光强度。
四、数据处理
1.绘制标准曲线。以荧光强度和溶液浓度作图,绘制标准曲线。
2.求出未知样浓度。由未知样品的荧光强度在工作曲线上求出未知样浓度。
五、思考题
1.解释为什么荧光强度的测量要在与激发辐射成直角的方向?
2.叙述如何绘制荧光发射光谱。
实验五 荧光光度法测定维生素B2的含量
一、实验目的
(1)学习荧光光度法测定多维葡萄糖粉中维生素B2的分析原理。
(2)掌握970CRT荧光光度计的操作技术。
二、实验原理
维生素B2,又叫核黄素,是橘黄色无臭的针状结晶。维生素B2易溶于水而不溶于乙醚等有机溶剂。在中性或酸性溶液中稳定,光照易分解,对热稳定。维生素B2水溶液在430~440nm蓝光或紫外光照射下会发生绿色荧光,荧光峰在535nm,在pH 6~7的溶液中荧光强度最大,在pH 11的碱性溶液中荧光消失。
由于维生素B2在碱性溶液中经光线照射,会发生光分解而转化为光黄素,后者的荧光比核黄素的荧光强的多。因此,测量维生素B2的荧光时,溶液要控制在酸性范围内,且须在避光条件下进行。
三、仪器与试剂
970CRT荧光分光光度计。
10μg/mL维生素B2标准溶液:准确称取10.0mg维生素B2,用热蒸馏水溶解后,转入1L棕色容量瓶中,冷却后加蒸馏水至标线,摇匀,置于暗处保存。
冰乙酸(AR);维生素B2试样溶液。
四、实验步骤
(1)标准曲线的绘制
于6只50mL容量瓶中,分别加入10μg/mL维生素B2标准溶液0.50、1.00、1.50、2.00、2.50、3.00mL,再各加入冰乙酸2.0mL,加水至标线,摇匀。在970 CRT荧光分光光度计上,用1cm荧光比色皿于激发波长440nm,发射波长540nm处,测量标准系列溶液的荧光强度。
(2)维生素B2样品浓度的测定
样品稀释后在相同的测量条件下,测量其荧光强度。平行测定三次。
五、实验记录及数据处理
以相对荧光强度为纵坐标,维生素B2的质量为横坐标绘制标准曲线。从标准曲线上查出待测试液中维生素B2的质量,并计算出维生素B2试样的浓度。
六、思考题
1.试解释荧光光度法较吸收光度法灵敏度高的原因。
2.维生素B2在pH=6~7时最强,本实验为何在酸性溶液中测定?
实验六 溴化钾压片法测绘苯甲酸的红外吸收光谱
(一) 实验目的
1.了解红外光谱测定的基本原理及固体和液体的常用制样方法
2.掌握KBr压片法的操作技能
3.解析红外光谱谱图
(二) 实验原理
将连续改变频率的红外光照射样品时,样品分子中某个基团的振动频率和外界红外辐射的频率一致,且分子的偶极距发生了改变,才产生红外吸收。通过红外光照射前后,在一些波长范围内变弱(被吸收),在另一些范围内则较强(不吸收),由光学信号转换成数字信号得到有机化合物谱图。
红外光谱分为三个区域:
近红外(0.75~2.5 mm,13330~4000 cm-1)
中红外(2.5~15.4 mm,4000 ~650 cm-1)
远红外(15.4~830 mm,650~12 cm-1)
有机物大部分基团的振动频率出现在2.5~25μm(4000~400 cm-1)的中红外区,因此红外光谱通常指中红外光谱。
气体、液体、固体样均可以测定,测定所需样品量少(mg级),不破坏样品,可以回收。
(三)仪器及试剂
Magna FT-IR Nicolet 550(Ⅱ)
FW-4型压片机
溴化钾粉末
苯甲酸
(四) 制样及测试
按一定比例取少量干燥的溴化钾与苯甲酸混合,研成粉末后装入压片机模具中,压片。将窗片置于红外附件上进行测试。
(五) 光谱谱图解析
1.对样品纯度、来源、元素分析及其他物理性质、谱学性质等方面的了解。
2.初步分析特征基团频率、特征宽强峰、倍频(泛频)及合频特征峰。
3.初步确定为某类化合物后,与标准谱图核对。
(六) 问题
1.影响红外光谱吸收峰位变化的主要因素是什么?
2.测试前,样品需经过哪些预处理?为什么?
3.苯环的取代类型有哪些?请说明相应的特征吸收峰位。
实验七 水样的pH值测定
一、实验目的
1.学习pHS-3C型酸度计的使用方法。
2.了解电位法测定水的pH值的原理和方法。
二 实验原理
在日常生活和工农业生产中,所用水的质量都有一定标准。在进行水质检验中,水的pH值是重要检验项目之一,如生活饮用水pH要求为6.5~8.5。低压锅炉水要求pH为10~12。电子工业、实验试剂配制则需要中性的高纯水。
现在测量水的pH值比较精确的方法是电位法,该法是将玻璃电极(指示电极)、饱和甘汞电极(参比电极)与待测试液组成原电池,用酸度计(一种精密电位差计)测量其电势。原电池用下式表示:
Ag|AgCl(s)|HCl(0.1mol·L-1)|玻璃膜|试液溶液(xmol·L-1)║KCl(饱合)|Hg2Cl2(s)|Hg
玻璃电极 被测溶液 甘汞电极
玻璃电极为负极,饱和甘汞电极为正极。在一定条件下,电池的电动势E与pH为直线函数关系(推导过程从略)。
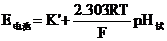
由上式看出,求出E电池和K,即可知道试液的pH值。E电池可通过测量求得,而K’是由内外参比电极及难于计算的不对称电位和液接电位所决定的常数,很难求得。在实际测量时,选用和待测试液pH值相似的、已知pH值的标准缓冲溶液在pH计上进行校正(这个过程叫定位),即测量pH是采用相对方法:
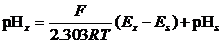
通过以上步骤,可在酸度计上直接读出试液的pH值。玻璃电极的响应斜率与温度有关,在一定的温度下应该是定值,25℃时玻璃电极的理论响应斜率为0.0591。但是玻璃电极由于制作工艺等的差异,每个pH玻璃电极其斜率可能不同,须用实验方法来测定。一支电极应使用两种不同pH值的标准pH缓冲溶液进行校正,两种缓冲溶液定位的pH值误差应在0.05之内。
三 仪器及试剂
1.仪器
pHS-3C酸度计一台,
E-201-C9型复合pH电极一支,(或231型玻璃电极和232型饱和甘汞电极各一支),
50mL烧杯7只。
2.试剂
①pH=4.00标准缓冲溶液(20℃):称取115±5℃下烘干2~3小时的一级纯邻苯二甲酸氢钾(KHC8H4O4)10.12g溶于不含CO2的蒸馏水中,在容量瓶中稀释至1000mL,贮于塑料瓶中。
②pH=6.86标准缓冲溶液(20℃):称取一级纯磷酸二氢钾(KH2PO4)3.39g和磷酸氢二钠(Na2HPO4)3.53g,将它们溶于不含CO2的蒸馏水中,在容量瓶中稀释至1000mL,贮于塑料瓶中。
③pH =9.18标准缓冲溶液(20℃):称取一级纯硼砂(Na2B4O7·10H2O)3.80g,将它溶于不含CO2的蒸馏水中,在容量瓶中稀释至1000mL,贮于塑料瓶中。
以上标准溶液也可用市售袋装缓冲溶液试剂直接配制。它们能稳定两个月,其pH值随温度不同稍有差异,见表1-2。
四 实验步骤
按照pHS-3C酸度计使用说明书进行操作。
1.酸度计的校正
(1)选用仪器的“pH”档;调节温度补偿,达到溶液温度值;
(2)把用蒸馏水清洗过的电极插入pH=6.86标准缓冲溶液中;
(3)调节定位,使仪器显示读数与该缓冲溶液当时温度下的pH相一致;
(4)用蒸馏水清洗电极,用滤纸吸干,再插入pH=4.00的标准缓冲溶液(若待测液是酸性)或pH=9.18的标准缓冲溶液(若待测液是碱性)中,
(5)调节使仪器显示读数与该缓冲液当时温度下的pH一致,仪器完成校正。
2.溶液pH的测定。
当被测溶液与标定溶液温度相同时,用蒸馏水缓缓淋洗两电极3~5次,再用滤纸吸干,然后将它们插入装有25~50mL被测溶液的烧杯中,电极浸入水中,摇动烧杯、使溶液均匀,待读数稳定后.读取pH;用蒸馏水清洗电极,滤纸吸干。
3.测量水样:分别测定4个不同水样的pH,每个水样应重复测定三次。(注意,应根据水样的pH,选择相应的标准pH缓冲溶液对仪器定位)
4.测量完毕后,放开测量开关,关上电源开关,拔掉电源,清洗电极,复合电极用3 mol·L-1KCl溶液浸泡。
五 数据处理
记录数据,填入下表中
表1-1 实验数据记录表
| 水样 |
水样1 |
水样2 |
水样3 |
水样4 |
| 测定次数 |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
| pH |
|
|
|
|
|
|
|
|
|
|
|
|
| 平均pH |
|
|
|
|
六 思考题
1.电位法测定水样的pH值的原理是什么?
2.酸度计为什么要用已知pH值的标准缓冲溶液校正?校正时应注意哪些问题?
3.何谓指示电极、参比电极?
实验八 离子选择性电极测定饮用水中的氟
一、实验目的
掌握直接电位法的测定原理及实验方法,并学会正确使用氟离子选择性电极和离子计的使用方法。
二、实验原理
饮用水中的氟含量的高低对人体的健康有一定的影响,氟的含量过低易得龋齿,过高则会发生氟中毒现象,适宜的含量为0.5毫克/升左右。
目前测定氟的方法有比色法和电位法。前者的测量范围较宽,但干扰因素多,往往要对试样进行预处理,后者的测量范围虽然范围不如前者宽,但一般能满足大多数水质分析的要求,而且操作简便,干扰少,样品一般不必进行预处理。因此,现在电位法测定氟离子以成为常规的分析方法。
本实验中应用氟离子选择性电极、饱和甘汞电极(SCE)和待测试液组成原电池。测量的电池电动势E与氟离子活度符合能斯特方程:
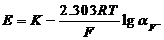
其中K、R、F均为常数,若在试液中加入适量的惰性电解质(如硝酸钠等),使离子强度保持不变,即使离子的活度系数为一常数,则上式中的氟离子活度可用浓度[F-]代替。25OC时上式可写作:
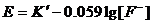
可见,电动势E与lg[F-]成线性关系。因此,只要作出E对lg[F-]的标准曲线,即可由水样测得的E,从标准曲线求得水样中的氟离子浓度。
三、仪器与试剂
1.仪器:
PXSJ-226离子计,CSB-F-1型氟离子选择性电极,饱和甘汞电极,磁力搅拌器
容量瓶 100mL 7个
移液管 50mL 1支
吸量管 0.5mL 1支 、10mL 2支
聚乙烯烧杯 100mL 7个
2.试剂:
氟化钠标准溶液,0.100 mol·L-1:称取4.1988g氟化钠(G.R),以去离子水溶解并稀释至1升,摇匀,储于聚乙烯瓶中备用。
总离子强度调节缓冲液(TISAB):取29克硝酸钠和0.2克二水合柠檬酸钠,溶于50毫升1:1(体积)的醋酸与50毫升5mol·L-1的氢氧化钠的缓冲溶液中,测量该溶液的pH,若不在5.0~5.5内,可用5mol·L-1的氢氧化钠和6mol·L-1的盐酸调节至所需范围。
四、实验步骤
1.调整仪器:
氟电极接离子计的负端,饱和甘汞电极接离子计的正端,仪器连接好电源后,打开电源开关,仪器即显示“PXSJ-226型离子计”等字样,稍等片刻,仪器自动进入起始状态,仪器必须开机预热0.5小时后方可进行测量。
注意:
①测量前需用电阻在3MΩ以上的去离子水浸泡活化一小时以上,当测得其在纯水中的毫伏数大于300mV时,便可用于测量。
②测量时,单晶薄膜上不可附有气泡,以免干扰读数。
③溶液的搅拌速度应缓慢、稳定。
2.标准曲线法:
(1)系列标准溶液的配制
用吸量管取10毫升0.100 mol·L-1氟化钠标准溶液,和10毫升TISAB溶液,在100毫升容量瓶内用去离子水稀释至刻度,摇匀。并用逐级稀释法配制成浓度为10-2、10-3、10-4、10-5、10-6mol·L-1的氟化钠系列标准溶液。逐级稀释时,只需加入9毫升TISAB溶液。
(2)标准曲线的绘制
将上述标准溶液依次倒入小聚乙烯塑料烧杯中(浸没电极即可),用滤纸吸去悬挂在电极上的水滴,插入氟离子选择电极和饱和甘汞电极,连接线路,放入搅拌子,开动搅拌器,由稀至浓分别测量标准溶液的电位值,待电位值稳定后读取读数。每次测定前用被测试液清洗电极、烧杯以及搅拌子。标准溶液测量完毕后将电极用蒸馏水清洗直至测得电位值300mV左右待用。
(3)饮用水样的测定:
用移液管移取50毫升饮用水样于100毫升容量瓶中,加入10毫升TISAB溶液,用去离子水稀释至刻度,摇匀。
清洗氟离子选择性电极,使其在纯水中测得的电动势在300毫伏。
将清洗后的电极用滤纸吸去悬挂着的水滴,插入盛有上述未知水样的烧杯中,搅拌数分钟,读取稳定的电动势。
五、数据及处理
记录对氟化钠系列标准溶液所测得的电动势E,并在坐标纸上作E对pF的标准曲线。
| 氟离子浓度 (mol·L-1) |
10-2 |
10-3 |
10-4 |
10-5 |
10-6 |
| pF |
2 |
3 |
4 |
5 |
6 |
| E(mV) |
|
|
|
|
|
记录未知试样溶液的电动势,并由标准曲线查得未知试样溶液中的氟离子浓度[F-],由下式计算饮用水中的氟离子含量。
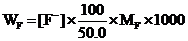
式中WF为每升饮用水样中氟离子的毫克数,设MF为氟的原子量。
六、思考题
1.氟离子选择性电极使用时应注意哪些问题?
2. TISAB的组成是什么?它在测量中起的作用是什么?
3.溶液的酸度对测定的影响如何?
实验九 库仑滴定法测定微量砷
[目的要求]
1、了解恒电流库仑法的基本原理。
2、掌握库仑滴定的基本原理和实验操作。
3、学习恒电流库仑滴定中终点指示法。
[基本原理]
库仑滴定法是建立在控制电流电解过程基础上的一种相当准确而灵敏的分析方法,可用于微量分析及痕量物质的测定。与待测物质起定量反应的“滴定剂”由恒电流电解在试液内部产生。库仑滴定终点借指示剂或电化学方法指示。按法拉第定律算出反应中消耗“滴定剂”的量,从而计算出砷的含量。
本实验用双铂片电极在恒定电流下进行电解,在铂阳极上KI中的I-可以氧化成I2。
在阳极 2I-→I2+2e
在阴极 2H++2e→H2↑
在阳极上析出的I2是个氧化剂,可以氧化溶液中的As(III),此化学反应为:
I2+AsO33-+H2O→AsO43-+2I-+2H+
滴定终点可以用淀粉的方法指示,即产生过量的碘时,能使有淀粉的溶液出现蓝色。也可用电流—上升的方法(死停法),即终点出现电流的突跃。
滴定中所消耗I2的量,可以从电解析出I2所消耗的电量来计算,电量Q可以由电解时恒定电流I和电解时间t来求得:Q=I×t (安培×秒)
本实验中,电量可以从KLT-1型通用库仑仪的数码管上直接读出。
砷的含量可由下式求得:
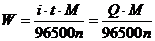
式中M为砷的原子量74.92,n为砷的电子转移数。
I2与AsO33-的反应是可逆的,当酸度在4 mol/L以上时,反应定量向左进行,即H2AsO4氧化I-;当pH>9时,I2发生岐化反应,从而影响反应的计量关系。故在本实验中采用NaH2PO4-NaOH缓冲体系来维持电解液的pH在7~8之间,使反应定量地向右进行。即I2定量的氧化H3AsO3。水中溶解的氧也可以氧化I-为I2,从而使结果偏低。故在标准度要求较高的滴定中,须要采取除氧措施。为了避免阴极上产生的H2的还原作用,应当采用隔离装置。
[仪器与试剂]
KLT-1型通用库仑仪;10 mL量筒;0.5 mL、5 mL移液管
试剂:(1)磷酸缓冲溶液:称取7.8 g NaH2PO4·2H2O和2 g NaOH,用去离子水溶解并稀释至250 mL(0.2 mol/L NaH2PO4;0.2 mol/L NaOH)。
(2)0.2 mol/L碘化钾溶液:称取8.3 g KI,溶于250 mL去离子水中即得。
(3)砷标准溶液:准确称取0.6600 g As2O3,以少量去离子水润湿,加入NaOH溶液搅拌溶解,稀释至80~90 mL。用少量H3PO4中和至溶液近于pH 7,然后转移至100 mL容量瓶中稀释至刻度,摇匀。此溶液浓度为砷5.00 mg/mL,使用时可进一步稀至500μg/mL。
[实验步骤]
1、调好通用库仑仪;
2、开启电源开关预热半个小时;
3、取10 mL 0.2 mol/L KI,10 mL 0.2 mol/L磷酸缓冲溶液,放于电解池中,加入20 mL蒸馏水,加入含砷水样5.00 mL,将电极全部浸没在溶液中。
4、终点指示选择电流—上升
5.按下电解按钮,灯灭,开始电解。数码管上开始记录毫库仑数。
6、电解完毕后,记下所消耗的毫库仑数。
7、再在此电解液中加入5.00 mL含砷水样,再做一次电解得到第二个毫库仑数,如此重复4次,得到4个毫库仑数。
8、舍去第一次的数据,取后三个的平均值,计算水样中的As量。以As mg/mL,或As2O3mg/mL表示。
[数据处理]
| 电 解 次 数 |
样 品 量 |
电 解 电 流 |
库 仑 数 |
| 1 2 3 4 |
|
|
|
[思考题]
1、0.1安培电流通过氰化亚铜溶液2小时,在阴极上析出0.4500 g铜,试求此电解池的电流效率。
2、库仑滴定的基本要求是什么?双铂电极为什么能指示终点?
实验十阳极溶出伏安法测定水中微量镉
一、实验目的
1:熟悉溶出伏安法的基本原理。学会阳极溶出伏安法测定水中微量镉的方法。
2:掌握LK1100电化学分析仪的操作方法。
二、方法原理
溶出伏安法的测定包含两个基本过程。即首先将工作电极控制在某一条件下,使被测定物质在电极上富集,然后施加线性变化电压于工作电极上,使被测物质溶出,同时记录电流与电极电位的关系曲线,根据溶出峰电流的大小来确定被测定物质的含量。
1电解富集(-1.0V, 富集时间t, 工作电极的表面积s,搅拌器的速度V)
Cd2++ 2e- + Hg = Cd(Hg)
2溶出测定(-1.0v→-0.2v)
本法使用汞膜电极为工作电极,铂电极为辅助电极,甘汞电极为参比电极。在被测物质所加电压下富集时,汞与被测物质在工作电极的表面上形成汞齐,然后在反向电位扫描时,被测物质从汞中“溶出”,而产生“溶出”电流峰。在KCl支持电解质中,当电极电位控制为-1.0v
时,Cd2+在工作电极上富集形成汞齐膜,然后当阳极化扫描至-0.2v时,可得到清晰的溶出电流峰。镉的波峰电位约为-0.6v左右。
三、仪器和试剂
1:LK1100 电化学分析仪,天津兰力科
2:汞膜电极作工作电极,甘汞电极作参比电极及铂辅助电极组成三电极系统。
3:10ugmL-1镉离子标准溶液、
4:1mol/L KCl溶液
5:10-3mol/LHgcl2
四、实验步骤
1:配制试液:移取水样25.00ml置于100ml烧杯中,分别加入1mol/LKCl
溶液5ml,10-3 mol/LHgcl2溶液5ml,少许Na2SO3(s)。
2:将未添加Cd2+标准溶液的水样置电解池中,放入清洁的搅拌磁子,插入处理好的电极系统。
3:打开仪器预热20分钟,打开电脑,打开LK1100电化学分析仪操作界面。
4:选择方法,溶出伏安法→差分脉冲溶出伏安法
5:设置参数,
6:实施实验:
(1)用标准加入法测定水样两次,量出hx1和 hx2 , 计算hx的平均值(2)加入Cd 2+
标准溶液10ug mL-1 200uL 同样测定两次, 量出H1和 H2,计算H的平均值.
五、数据处理
1:列表记录所测定的实验结果。
2:取两次测定的平均峰高,按公式计算水样中Cd2+的浓度。
Cs Vs hx
Cx = ______________________________
H(Vx +Vs) - hxVx
式中: hx:测得水样的峰电流高度两次的平均值(A)
H:水样加入标准溶液后测得总高度两次的平均值(A)。
Cs:标准溶液的浓度。10 ug mL-1
Vs:标准溶液的体积。200 uL
Vx:取水样的体积。25.00 mL
六、思考题
1:阳极溶出伏安法为什么有较高的灵敏度。
2:阳极溶出伏安法为什么必须保持相同的实验条件。
实验十一 液相色谱法测定污染水样中的甲苯
一、实验目的
1.液相色谱仪的整套装置、工作原理、工作流程;会操作和使用化学工作站。
2.掌握外标法测定甲苯的实验方法。
二、 实验原理
液相色谱法就是同一时刻进入色谱柱中的各组分,由于在流动相和固定相之间溶解、吸附、渗透或离子交换等作用的不同,随流动相在色谱柱中运行时,在两相间进行反复多次(103~106次)地分配过程,使得原来分配系数具有微小差别的各组分,产生了保留能力明显差异的效果,进而各组分在色谱柱中的移动速度就不同,经过一定长度的色谱柱后,彼此分离开来,最后按顺序流出色谱柱而进入信号检测器,在记录仪上或色谱数据机上显示出各组分的色谱行为和谱峰数值。测定各组分在色谱图上的保留时间(或保留距离),可直接进行组分的定性;测量各峰的峰面积,即可作为定量测定的参数,采用工作曲线法(即外标法)测定相应组分的含量。
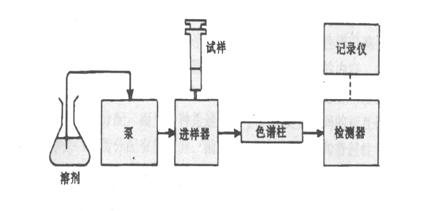
液相色谱仪工作原理图
高效液相色谱仪是实现液相色谱分离分析过程的装置,如上图所示。贮液器中存贮的载液(用作流动相的液体常需除气)经过过滤后由高压泵输送到色谱柱入口(当采用梯度洗脱时,一般需用双泵系统来完成输送)。样品由进样器注入载液系统,而后送到色谱柱进行分离。分离后的组分由检测器检测,输出信号供给记录仪或数据处理装置。如果需收集馏分作进一步分析,则在色谱柱出口将样品馏分收集起来,对于非破坏型检测器,可直接收集通过检测器后的流出液。其中输液泵,色谱柱及检测器是仪器的关键部件。
三、仪器与试剂
1. 色谱仪:奥泰高效液相色谱仪。
色谱柱;C18 5 μm 4.6×200mm
流动相:甲醇:水=90:10 流速:1.0 mL/min
检测器:UV-254nm 温度:室温
数据处理器:Chromeleon色谱工作站。
苯、甲苯的标准溶液浓度:0.125mg/mL、0.25 mg/mL、0.50 mg/mL、0.75 mg/mL、1 mg/mL
四、实验步骤
1、分别精密称取适量的苯、甲苯标准品于100 mL容量瓶中,用甲醇溶解,并稀释至刻度作为标准溶液。
2、取适量苯、甲苯混合样品于100 mL容量瓶中并用甲醇溶解至刻度作为未知样。
3、用超纯水(经0.22 μm滤膜过滤)和甲醇(色谱纯)配制成比例为90:10的混合溶液作为流动相。
4、依次打开色谱工作站、主机、脱气器及紫外检测器的电源开关。
5、调整检测器波长在254nm处,观察色谱工作站中色谱图基线情况。
7、待基线平稳后,用微量进样器分别取20 μL标准溶液,由进样阀进样,同时记录色谱图和保留时间。
8、取未知样品20 μL进样,记录色谱图和保留时间。
五、数据处理
1、根据得到的标准曲线色谱图与保留时间,在未知样品谱图中确定每个峰的归属。
| 标准品 |
苯 |
甲苯 |
| 保留时间(min) |
|
|
| 未知混合样品 |
1号峰 |
2号峰 |
| 保留时间(min) |
|
|
| 各峰对应的组分 |
|
|
2、通过相应的色谱峰峰面积计算出标准曲线的回归方程和线性相关系数
| 标准样 |
浓度1 |
浓度2 |
浓度3 |
浓度4 |
浓度5 |
| 各组分峰面积 |
苯 |
|
苯 |
|
苯 |
|
苯 |
|
苯 |
|
| 甲苯 |
|
甲苯 |
|
甲苯 |
|
甲苯 |
|
甲苯 |
|
| 标准曲线 |
苯 |
|
相关系数 |
|
| 甲苯 |
|
|
3、计算出未知样品中待测组分的含量。
4、打印出相应的色谱图。
六、注意事项
1、要注意观察泵的压力值,如有异常,要及时停泵,
2、使用微量注射器时要注意取量的准确性,
3、注射样品至进样阀时,要将注射器推到阀的根部。
实验十二气相色谱法检测白酒中甲醇的条件选择及测定
我国有着悠久的饮酒文化和历史,随着人民生活水平的提高,白酒的消费也不断增加,人们对白酒的品质和安全也更加关心。白酒中的甲醇作为一种有毒物质对人们的健康存在威胁,必须限定其含量。
1. 仪器与试剂
1. 1仪器
气相色谱仪( Agilent 6890N) :配有自动进样器( Agilent 7890B)、氢火焰离子化检测器( FID) ;色谱柱:毛细管柱HP-INNOWAX,长30 m,内径0. 32 mm,内膜0. 25μm;全玻璃蒸馏装置:500 mL;容量瓶: 100 mL;卡口瓶;可调式移液器。
1. 2试剂
无水甲醇(山东,色谱纯),作标样用;乙腈(苏州,色谱纯),作溶剂用;蒸馏水。
酒样
2实验测定条件
开机:打开电源之前先检查各气路、电路的连接是否正确,并进行下列条件设置:
选择进样量应为1μL
汽化室的温度为220℃;
选择检测器的温度为250℃;
柱温为40℃;
选择载气流量为N2为载气流速1. 0 mL /min
氢气流速为30 mL /min。
以上条件设定完成后,先打开色谱仪主机总电源(开关位于右测下方),开启载气钢瓶阀,调节恒压输出。再打开电源,开启计算机。
3.标准曲线的建立
3. 1标准储备液的配制
先在100 mL容量瓶中装少量色谱乙腈,再精密量取1 mL色谱甲醇置于此容量瓶中,用色谱乙腈定容至刻度,制0. 0079 g /mL的甲醇储备液。
3. 2标准使用液的配制
准确吸取甲醇标准储备液0、1、2、4、6、8、10 mL,分别置于10 mL的容量瓶中,用色谱乙腈定容至刻度,振荡,混匀,备用。
3. 3标准工作曲线的绘制
准确吸取1μL标准使用液,每个浓度平行测定5次取平均值。测得标准甲醇使用液在最佳色谱条件下,其峰面积和甲醇浓度之间的关系。以峰面积对甲醇浓度绘制标准曲线样品的测定,进样1.0μL,测得样品色谱图,由化学工作站自动计算出样品中各组分的含量。
4.结果处理
5.注意事项
(1)开机前先开载气,实验结束后柱温降至所需温度后关闭载气。
(2)关闭空气压缩机时必须放气。
